2023: Volume 4, Issue 3
Past Issues
 Abstract
Abstract  PDF
PDFIn Silico Discovery and ADMET Pharmacokinetic of Novel Pyrimidinic Selenoureas as Selective Breast Carcinoma Cells (MCF-7) Inhibitors
Yusuf Isyaku1,*, Adamu Uzairu2, Aliyu Mohammed Ja'o1
1Department of Chemical Sciences, Federal University of Kashere, Gombe State, Nigeria
2Department of Chemistry, Ahmadu Bello University, Zaria, Kaduna State, Nigeria
*Corresponding Author: Yusuf Isyaku, Department of Chemical Sciences, Federal University of Kashere, Gombe State, Nigeria; Email: [email protected]
Received Date: July 14, 2023
Publication Date: August 1, 2023
Citation: Isyaku Y, et al. (2023). In Silico Discovery and ADMET Pharmacokinetic of Novel Pyrimidinic Selenoureas as Selective Breast Carcinoma Cells (MCF-7) Inhibitors. Clin Res. 4(3):16.
Copyright: Isyaku Y, et al. © (2023).
ABSTRACT
Pyrimidine is one of aromatic heterocyclic class of organic compounds that’s similar to pyridine. It’s found in the nucleic acids DNA and RNA. Novel pyrimidinic selenoureas were reported to have a remarkable inhibitory activity against breast carcinoma cells (MCF-7). With the help of computer-aided drug design techniques one of these of compounds was further optimized to design three other derivatives with more potency than the previous and also more potent than many anti-breast cancer drugs. The main aim of this study was to design more potent pyrimidinic selenoureas derivatives and compare them with the standard anti-breast cancer drugs. An optimization method of structure-based drug design was employed. Two compounds of novel pyrimidinic selenoureas were reported in which the first compound was selected and docked with the ERBB2 receptor tyrosine kinase (PDB ID: 2A91), it was then modified to design three (3) derivatives. The receptors were later docked with seven (7) different anti-breast cancer drugs approved by American Cancer Society (such as, Capecitabine, Cisplatin, Curcumin, Paclitaxel, Ixabepilone, Doxorubicin and Vinorelbine) to record their potency and later compared with the designed compounds. An ADMET pharmacokinetic study was carried out on the designed compounds to investigate their drug-likeness. In the result, all the designed compounds were found to be more potent than the template, in which compound a1 and a2 (with moldock score of -148.456 and -153.725) were found to be more potent than Capecitabine, Cisplatin, Curcumin, Doxorubicin and Vinorelbine (moldock score; -134.953, -43.889, -148.290, -106.187 and -134.986), but less active than Paclitaxel and Ixabepilone (with moldock scores of -154.135 and -157.093), with compound a3 (moldock score; -161.583) recorded the highest potency which is more potent than all the listed drugs, and also the designed compounds were found to have good pharmacokinetic profiles. Conclusively, three other derivatives of novel pyrimidinic selenoureas were designed and found to be more potent than the template and many anti-breast cancer drugs. The compounds should be further synthesized for their excellent activities and good pharmacokinetic parameters.
Keywords: In silico Discovery, Novel pyrimidinic selenoureas, MCF-7 inhibitors, ADMET Pharmacokinetic
INTRODUCTION
The dramatic increase of death caused by breast cancer is usually due to lack of various types of therapies [1]. Pyrimidine is one of aromatic heterocyclic class of organic compounds that’s similar to pyridine [2]. It’s found in the nucleic acids DNA and RNA (Pyrimidine, 2021). Pyrimidine compounds were found to be significances in the field of medicine, such as anticancer [3], antimicrobial [4], antimalarial [5] among other.
Organoseleniums are renounced for their antioxidant activity as well as their capacity to imitate selenoenzyme Glutathione Peroxidase (GPx-like activity) [6].
Also an existing evidence indicated that compounds of selenium (Se) had potential in cancer chemotherapy; thus, they are much renounced in inhibiting cell proliferation and lead to death in human cancer cells via apoptosis [7,8].
Computer-aided drug design equipped us with the knowledge of receptor-ligand complex which give an insight for structural modified to design more potent drug candidate. This can be achieved through different methods, here we used the method of optimization where a compound was chosen and modified to get more potent derivatives. The aim of the research was to design more potent anti-breast cancer compounds via in silico technique of drug discovery.
MATERIALS AND METHOD
Preparation of Ligand
From the work of Barbosa, et al. (2018) [9] compound 8a was selected and modified because of it greater potency against MCF-7 and HeLa cell lines of breast cancer. The compound was drawn using Chemdraw Ultra version 12.0 software and optimized using spartan 14 software where it was later converted to PDB file (Ibrahim et al., 2018) [10] and then docked with the ERBB2 receptor tyrosine kinase (PDB ID: 2A91) using molegro software. The result of the docking study was recorded and later compared with the results of its derivatives. Following the same procedure, the 2D structures of the three (3) designed compounds were drawn, optimized and then converted to Protein Data Bank (PDB) format. Molegro software was also used to perform the docking study [11].
Preparation of Receptor
From the protein databank website (www.rcsb.org) the 3D structures of ERBB2 receptor tyrosine kinase (PDB ID: 2A91) was downloaded. With the aid of molegro virtual docker software, the downloaded receptor was prepared by removing all ligands, heteroatoms and water molecules present in it. Subsequently, the prepared receptors alongside the prepared ligands were docked using docking wizard in a melogro software. Molegro Visualizer was then used to visualize the docking results. The prepared ligand and receptor are shown in Figures 1 and 2.

Figure 1: Structure of the prepared ligand (Scaffold).

Figure 2: Structure of the prepared receptor.
Optimization method for structure-based design
This refers to the optimization of known molecules through evaluating its proposed analogs within the binding cavity [12]. Discovery studio was used to visualize the receptor- ligand interactions in which different interactions such as H-bond and hydrophobic interactions formed between the Scaffold compound and the receptor (ERBB2 receptor tyrosine kinase) were studied. Based on the knowledge of these interactions, the designed compounds were proposed in which they were drawn, optimized, converted to PDB and later docked with the receptors to record their potency [13]. The receptors was later docked with seven (7) different anti-breast cancer drugs approved by American Cancer Society (such as, Capecitabine, Cisplatin, Curcumin, Paclitaxel, Ixabepilone, Doxorubicin and Vinorelbine) to record their potency and later compared with the designed compounds.
Theoretical prediction of ADME/T Parameters
A designed computer program known as ADMET Predictor is used to predict the pharmacokinetic parameters for a drug-likeness often referred to as “ADME/T” (Absorption, Distribution, Metabolism, Excretion/Elimination, and Toxicity) [14]. This study accounts for about 60% failures of the drug candidates. Being more potent and less toxic are not enough to qualify drug candidate, a good pharmacokinetic profile is very vital step in drug discovery. Therefore, to avoid waste of time and resources it is significantly important to examine the ADMET profile of designed compounds earlier. Thus, an online software known as swissADME was used to predicted the ADMET profiles of the designed compounds (a1 – a3) [15].
In 1997, Lipinski, et al. [16] proposed the "Rule of Five" which was made of four (4) ADMET parameters. At the then time, the Rule-of-Five was the "well-known rule-based filter" for examining whether a compound can be orally absorbed or not. The Rule of Five are; Molecular weight (MW), Number of hydrogen bond acceptors (HBAs), Number of hydrogen bond donors (HBDs), and Octanol/water partition coefficient (iLOGP). According to Rule of Five, a drug can be orally absorb only if it doesn’t cause more than one violation of the rules above. However, the Rules are not applicable to some complicated natural products [17,18], thus, the concept of QED (quantitative estimate of drug-likeness) comes into being which developed by Hopkins in 2012 [19] that brought about eight physicochemical parameters. These parameters are the Rules of Five and four(4) others; such as, number of aromatic rings (AROMs), molecular polar surface area (TPSA), number of alerts for undesirable substructures (ALERTs i.e. PAINS #alert and Brenk #alert) and number of rotatable bonds (ROTBs) [19]. The most flexible and adopted rules for drug-likeness is the concept of QED.
To examine the pharmacokinetic profile of the designed molecules, Chemdraw Ultra 12.0 was used to draw the chemical structure of the compounds. The molecules were imported into the website’s interface (http://swissadme.ch/). The SwissADME drug parameters were then generated [20].
Some of the ADME/T parameters and their recommended values are represented in Table 1.
Table 1: Some ADME/T parameters and their recommended values.
|
Parameter |
Default range |
|
Molecular weight (MW) |
50–500 |
|
octanol/water partition coefficien (iLOGP) |
-2–10 |
|
Topological Polar Surface Area (TPSA) |
20–130 |
|
Number of H-Bond acceptors (HBA) |
0–10 |
|
Number of H-bond Donors (HBD) |
0–5 |
|
Rotatable bonds (RB) |
0–5 |
|
Number of heavy atoms (nHA) |
15–50 |
RESULTS
Design
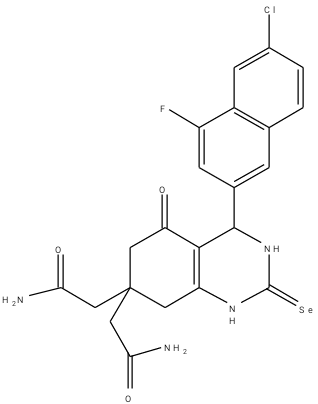
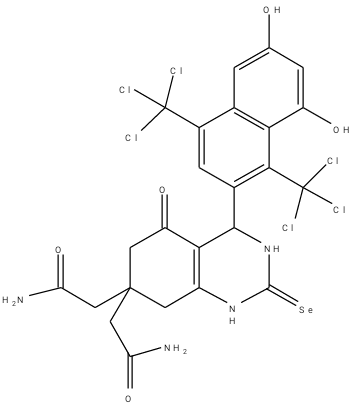
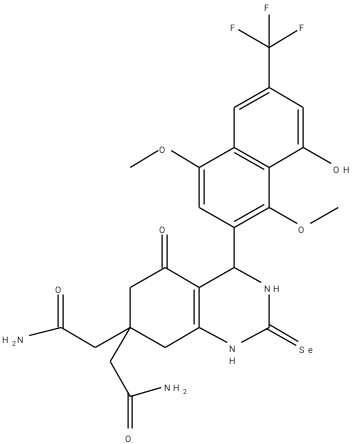
An optimization method of structure-based design was adopted in this work. From the work of Barbosa et al. (2018) [9] compound 8a was selected and modified in which three (3) other derivatives were designed. The optimization method of structure-based drug design involves the use of molecular docking results. Table 2 is the designed compounds and their moldock scores.
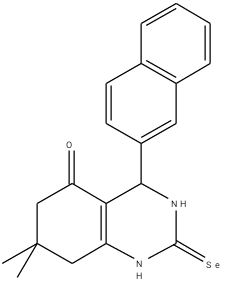
Figure 3: Chosen scaffold compound 8a (MolDock = -109.937)
Table 2: Structures of the designed compounds with their binding affinity.
|
Serial No. |
Structure |
MolDock Score |
|
a1 |
|
-148.465 |
|
a2 |
|
-153.725 |
|
a3 |
|
-161.583 |
The 3D and 2D docking interaction between the compound a3 and protein residuals from the receptor are shown in Figures 4 and 5.
Figure 4: 3D structure of receptor-ligand interaction for the compound a3.
Figure 5: 2D structure of receptor-ligand interaction for the compound a3.
ADMET Calculation
Table 3: Calculated ADME parameters of the designed compounds.
|
Compound |
MW |
iLOGP |
HBA |
HBD |
TPSA |
nAH |
PAINS #alerts |
Brenk #alerts |
|
a1 |
521.83 |
0 |
4 |
4 |
127.31 |
10 |
0 |
1 |
|
a2 |
670.75 |
0 |
7 |
5 |
147.54 |
10 |
0 |
2 |
|
a3 |
613.44 |
0 |
9 |
5 |
166 |
10 |
0 |
1 |
Molecular weight (MW), number of rotatable bonds (RB), number of hydrogen donors (HBD), number of hydrogen acceptors (HBA), Topological Polar Surface Area (TPSA), octanol/water partition coefficient (iLOGP), number of aromatic heavy atoms (nAH), Molar refractivity (MR), number of alerts for undesirable substructures/substructures (Brenk #alert and PAINS #alert).
DISCUSSION
Design
In this study, a structure-based drug design approach was employed to design new anti-breast cancer compounds with better activity beginning with the compound 8a (Chosen scaffold compound) with MolDock score of -109.937 as our lead compound (Figure 3) which was reported to have good anti-breast cancer activity. On the bases of the interaction between the compound and the receptor, a series of three (3) derivatives of the compound (Table 2) were designed through structural modification of the compound as understood from its docking analysis. All the proposed compounds were docked using Molegro Virtual Docker and their binding free energies were recorded. All the designed compounds were found to be more potent than the template, in which compound a1 and a2 (with moldock score of -148.456 and -153.725) were found to be more potent than the standard drugs Capecitabine, Cisplatin, Curcumin, Doxorubicin and Vinorelbine (moldock score; -134.953, -43.889, -148.290,-106.187 and -134.986), but less active than Paclitaxel and Ixabepilone (with moldock scores of -154.135 and -157.093), with compound a3 (moldock score; -161.583) recorded the highest potency which is more potent than all the listed drugs.
ADME/T Study
The results of pharmacokinetic study unveiled that the designed compounds (a1, a2 and a3) were found to have good pharmacokinetic profile. That is to say, the physicochemical parameters (MW, iLOGP, HBA, HBD, TPSA, nAH, PAINS #alert and Brenk #alert) are within the recommended values (Table 1). From the results of the ADMET study (Table 3), it can be observed that there are few violations of the recorded rules, i.e. as according with Lipinski's Rule of Five/concept of QED which indicated they are within the acceptable profile. However, there was no alert of PAINS which signified that the molecules are quite specific. Thus, it can now be said that, the designed anti-breast cancer agents possess good pharmacokinetic profiles.
CONCLUSION
Conclusively, using an optimization method of structure-based design, three other derivatives of novel pyrimidinic selenoureas were designed and found to be more potent than the template and many anti-breast cancer drugs. The compounds should be further synthesized for their excellent activities and good pharmacokinetic profiles.
REFERENCES
- Al-Gazali HOM, Kzar HH, Wtwt MA, Al-Gazally ME. (2021). EV Targeting MCF-7 Breast Cancer Cell Lines Inhibit Both mTOR and HIF-1A: Molecular Docking Study. Med Legal Update. 21(1):1367-1373.
- Gilchrist TL. (1997). Heterocyclic chemistry, New York: Longmam.
- Kilic-Kurt Z, Ozmen N, Bakar-Ates F. (2020). Synthesis and anticancer activity of some pyrimidine derivatives with aryl urea moieties as apoptosis-inducing agents. Bioorganic Chem. 101:104028.
- Zhuang J. Ma S. (2020). Recent Development of Pyrimidine‐Containing Antimicrobial Agents. ChemMedChem. 15(20):1875-1886.
- Iman M, Davood A, Khamesipour A. (2020). Design of antimalarial agents based on pyrimidine derivatives as methionine aminopeptidase 1b inhibitor: Molecular docking, quantitative structure activity relationships, and molecular dynamics simulation studies. J Chinese Chem Soc. 67(5):880-890.
- Bhabak KP. Mugesh G. (2010). Functional mimics of glutathione peroxidase: bioinspired synthetic antioxidants, Acc Chem Res. 43: 1408–1419.
- Sinha R, El-Bayoumy K. (2004) Apoptosis is a Critical Cellular Event in Cancer Chemoprevention and Chemotherapy by Selenium Compounds, Curr Cancer Drug Targets. 4:13-28.
- Rikiishi H. (2007). Apoptotic cellular events for selenium compounds involved in cancer prevention. J Bioenerg Biomembr. 39:91-98.
- Barbosa FA, Siminski T, Canto RF, Almeida GM, Mota NS, Ourique F, et al. (2018). Novel pyrimidinic selenourea induces DNA damage, cell cycle arrest, and apoptosis in human breast carcinoma. Eur J Med Chem. 155:503-515.
- Ibrahim MT, Uzairu A, Shallangwa GA, Ibrahim A. (2018). Computational studies of some biscoumarin and biscoumarin thiourea derivatives as ⍺-glucosidase inhibitors. J Engineer Exact Sci. 4(2):0276-0285.
- Molegro A. (2011) MVD 5.0 Molegro virtual docker. DK-8000 Aarhus C, Denmark.
- Klebe G. (2000). Recent developments in structure-based drug design. J Mol Med. 78 (5):269-281.
- Arthur DE, Uzairu A. (2018). Molecular docking study and structure‐ based design of novel camptothecin analogues used as topoisomerase I inhibitor. J Chinese Chem Soc. 65(10):1160-1178.
- Singh DB, Gupta MK, Singh DV, Singh SK, Misra K, (2013). Docking and in silico ADMET studies of noraristeromycin, curcumin and its derivatives with Plasmodium falciparum SAH hydrolase: a molecular drug target against malaria. Interdiscip Sci. 5(1):1-12.
- Daina A, Michielin O, Zoete V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 7:42717.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 23(1-3):3-25.
- Ghose AK, Viswanadhan VN, Wendoloski JJ. (1999). A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Combo Chem. 1(1):55-68.
- Bhal SK, Kassam K, Peirson IG, Pearl GM. (2007). The rule of five revisited: Applying log D in place of log p in drug-likeness filters. Mol Pharm. 4(4):556-560.
- Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. (2012). Quantifying the chemical beauty of drugs. Nat Chem. 4(2):90.
- Mishra S, Dahima R. (2019). In vitro ADME studies of TUG-891, a GPR-120 Inhibitor using swiss ADME predictor. J Drug Deliv Ther. 9(2-s):366-369.