2023: Volume 4, Issue 2
Past Issues
 Abstract
Abstract  PDF
PDFProximal 15q11.2 Microduplication: A Case Report of Variable Expressivity
Gunce Basarir1,*, Nihal Olgac Dundar2, Irmak Erdogan1, Yasar Bekir Kutbay3, Pinar Gencpinar2
1University of Health Sciences, Tepecik Training and Research Hospital, Department of Pediatric Neurology, Izmir, Turkey
2Izmir Katip Celebi University, Faculty of Medicine, Department of Pediatric Neurology, Izmir, Turkey
3University of Health Sciences, Tepecik Training and Research Hospital, Genetic Diagnosis Center, İzmir, Turkey
*Corresponding Author: Gunce Basarir, University of Health Sciences, Tepecik Training and Research Hospital, Department of Pediatric Neurology, 35620, Konak, Izmir, Turkey; Tel: +905054024855; Email: [email protected]
Received Date: April 21, 2023
Publication Date: May 11, 2023
Citation: Basarir G, et al. (2023). Proximal 15q11.2 Microduplication: A Case Report of Variable Expressivity. Clin Res. 4(2):11.
Copyright: Basarir G, et al. © (2023).
ABSTRACT
The four highly conserved and non- imprinted genes (NIPA1, NIPA2, CYFIP1 and TUBGCP5) are located on the proximal 15q in the region between BP1 and BP2 which spans approximately 500 kb. Here, we report on a paternally inherited 15q11.2 breakpoint 1-2 microduplication carrier with epilepsy, near-normal neurodevelopment, mild intellectual disability and speech delay. The father had a normal phenotype, suggesting the variable expressivity. Proximal 15q breakpoint 1-2 region copy number variants may not warrant a clinical outcome since the phenotypic variability and low penetrance. We believe that greater understanding of the possible underlying molecular, genetic, and environmental modifying factors of the copy number variants in this susceptibility locus will aid in our clinical approach.
INTRODUCTION
The proximal long arm of chromosome 15 (15q) is known to be relatively susceptible to the non-allelic homologous recombination of low copy repeat elements at the breakpoint (BP) regions. (Gillentine & Schaaf, 2015) [1]. Among the six BP regions (BP1-BP6) on 15q, the BP1-BP3 region, defined as Prader-Willi/Angelman syndrome critical region, mediates deletions resulting in Prader-Willi and Angelman syndromes (PWS/AS) (Chamberlain & Lalande, 2010) [2]. Patients with PWS/AS type I deletions (BP1-BP3) exhibit a more severe neuropsychiatric phenotype compared to those with PWS/AS type II deletions (BP2-BP3), suggesting the BP1-BP2 region to be critical in neurodevelopmental processes (Butler, Bittel, Kibiryeva, Talebizadeh, & Thompson, 2004; Sahoo et al., 2007)[3,4].
The region spanning approximately 500 kilobases (kbs) between BP1 and BP2 contains four highly conserved and non-imprinted genes: NIPA1, NIPA2, CYFIP1, and TUBGCP5 (Chai et al., 2003) [5]. Several reports have linked microdeletions and microduplications of these four genes with behavioral problems, developmental delay, learning disabilities, seizures, and psychiatric problems including autism spectrum disorders (ASD), attention deficit/hyperactivity disorder (ADD/ADHD) (Doornbos et al., 2009; van der Zwaag et al., 2010; Burnside et al., 2011; von der Lippe, Rustad, Heimdal, & Rødningen, 2011; Abdelmoity, LePichon, Nyp, Soden, Daniel, & Yu, 2012; Vanlerberghe et al., 2015; Picinelli et al., 2016; Mohan et al., 2019) [6-13]. Recent studies also reported a more severe phenotype in the patients with microdeletions than those with microduplications involving these four genes (Burnside et al., 2011; Benítez-Burraco, Barcos-Martínez, Espejo-Portero, & Jiménez-Romero, 2017) [8,14]. Moreover, the diverse range of phenotypes, ranging from individuals with severe neuropsychiatric manifestations to asymptomatic carriers, suggest variable expressivity and incomplete penetrance for copy number variants (CNVs) of this highly unstable region (Benítez-Burraco, Barcos-Martínez, Espejo-Portero, & Jiménez-Romero, 2017) [14].
In this report, we present phenotypic features of a patient with 15q11.2 microduplication involving NIPA1, NIPA2, CYFIP1 and TUBGCP5 genes.
CLINICAL PRESENTATION
A 9-year-old boy was referred to the pediatric neurology clinics due to the generalized tonic-clonic status epilepticus. Brain magnetic resonance imaging, interictal electroencephalography (EEG) and detailed neurological examination of the patient were unremarkable at admission.
The patient was only child born to healthy, nonconsanguineous parents at full term by a caesarean section delivery due to previous uterine surgery. Perinatal history was unremarkable. The patient has had a history of generalized tonic seizures in the neonatal and infantile periods. He had achieved independent sitting and walking at the age of 11 and 20 months, respectively. A mild speech delay was also reported, since he was able to speak single words at the age of 20 months. He had no remarkable family history for neuropsychiatric problems, epilepsy and developmental delay. During the follow-up, bilateral temporo-occipital epileptic discharges were detected in one of the repeated interictal EEG recordings, while the remaining EEGs showed no abnormality. A near-normal neurodevelopment with a mild intellectual/learning disability was also noted at the subsequent visits. The patient is seizure-free for two years with sodium valproate anti-seizure medication. He still continues regular follow-up visits at pediatric neurology and pediatric psychiatry outpatient clinics.
GENETIC ANALYSIS
Chromosome microarray analysis was performed using the “Affymetrix GeneChip Array 350K” for copy number variants. The patient was found to have a 507-kb duplication involving 4 OMIM genes [TUBGCP5 (608147), CYFIP1 (606322), NIPA2 (608146), NIPA1 (608145)] at 15q11.2. The microarray analysis was reported as arr [hg19] 15q11.2(22,7-23,2)x3. The father was also found to have a 854-kb duplication at the same region. The microarray analysis was reported as arr [hg19] 15q11.2(22,7-23,6)x3. The mother’s microarray analysis was normal.
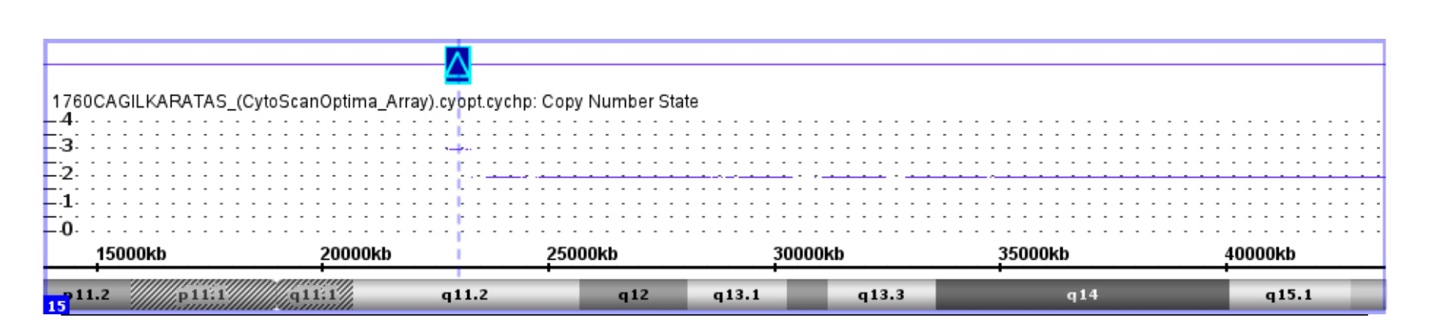
Figure 1: Visualization of the copy number variation in the BP1-BP2 region of 15q11.2
DISCUSSION AND CONCLUSION
Here in the present report, we describe phenotypic features of a patient with a paternally inherited microduplication of 507 kbs in the 15q11.2 BP1-BP2 region which encompasses four non-imprinted genes: NIPA1, NIPA2, CYFIP1, and TUBGCP5. These four genes have several functions in neuronal connectivity and axonal growth (Picinelli et al., 2016) [13]. NIPA1 and NIPA2 encode magnesium transporters in many tissues including brain (Goytain, Hines, El-Husseini, & Quamme, 2007; Goytain, Hines, & Quamme, 2008) [15,16]. Mutations in the NIPA1 gene have been linked to autosomal dominant hereditary spastic paraplegia (Reed et al., 2005; Goytain, Hines, El-Husseini, & Quamme, 2007) [16,18], while NIPA2 has been reported as a susceptible gene for childhood absence epilepsy (Jiang et al., 2012) [18]. CYFIP1 interacts with fragile-x mental retardation protein in neurons, playing a crucial role in fragile-X syndrome and ASD phenotypes (Nowicki et al., 2007) [19]. TUBGCP5, encoding a protein for cytoskeleton tubulin complex, has also been related to several neurobehavioral disorders (De Wolf, Brison, Devriendt, & Peeters, 2013) [11].
A wide range of phenotypes including normal phenotypes as well as neurodevelopmental delay, behavioral disturbances, ADHD, ASD, OCD, schizophrenia, epilepsy, anorexia, and mild dysmorphic features have been reported in individuals with microduplications located in the 15q11.2 BP1-BP2 region (van der Zwaag et al., 2010; Burnside et al., 2011; Abdelmoity, LePichon, Nyp, Soden, Daniel, & Yu, 2012; Kirov et al., 2012; Picinelli et al., 2016; Benítez-Burraco, Barcos-Martínez, Espejo-Portero, & Jiménez-Romero, 2017; Chang et al., 2019; Mohan et al., 2019) [8-10, 12-14, 20-22]. Our patient had epilepsy, mild developmental delay and intellectual/learning disability. The father of the patient had a normal phenotype although the microduplication was inherited paternally in our case, supporting the variable expressivity for CNVs of this region (Benítez-Burraco, Barcos-Martínez, Espejo-Portero, & Jiménez-Romero, 2017) [14]. Since the CNVs on this region may not be sufficient to cause a phenotype, secondary alterations may be needed to have phenotypic features (Burnside et al., 2011) [9].
Mohan et al (2019), [14] reported phenotypic features including developmental delay, dysmorphic features, ASD, and epilepsy/seizures in 159 of 215 (74%) 15q11.2 microduplication carrier individuals. Moreover, they noted the phenotypic variability and low penetrance in the families with 15q11.2 CNVs (Mohan et al., 2019) [14]. In another family report (a father and 3 male twin siblings) of microduplication in the 15q11.2 BP1-BP2 region , the father and fraternal twin were phenotypically normal, whereas the monozygotic twins had behavioral disturbances, intellectual disability and language delay, suggesting the variable penetrance (Benítez-Burraco, Barcos-Martínez, Espejo-Portero, & Jiménez Romero, 2017) [15]. A previous study revealed that microduplications on the BP1-BP2 region may also impact cerebral white matter microstructure. They found decreased fractional anisotropy in the duplication group when compared with the control group (Silva et al., 2019) [23]. More, previous reports indicate that microdeletions in 15q11.2 BP1-BP2 region may result in generalized epilepsy (Burnside et al., 2011; Jiang et al., 2012; Mohan et al., 2019) [9,14,19]. Epilepsy is known to be a less common phenotype in individuals with 15q11.2 BP1-BP2 microduplication than those with microdeletion of this region (Burnside et al., 2011; Mohan et al., 2019) [9,14]. However, our patient had seizures during neonatal-infantile periods and childhood.
To conclude, we reported on a paternally inherited 15q11.2 BP1-BP2 microduplication carrier with epilepsy, near-normal neurodevelopment, mild intellectual disability and speech delay. The father had a normal phenotype, suggesting the variable expressivity. Proximal 15q BP1-BP2 region CNVs may not warrant a clinical outcome since the phenotypic variability and low penetrance. We believe that greater understanding of the possible underlying molecular, genetic, and environmental modifying factors of the CNVs in this susceptibility locus will aid in our clinical approach.
REFERENCES
- Gillentine MA, Schaaf CP. (2015). The human clinical phenotypes of altered CHRNA7 copy number. Biochem Pharmacol. 97(4):352-362.
- Chamberlain SJ, Lalande M. (2010). Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13. Neurobiol Dis. 39(1):13-20.
- Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. (2004). Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. 113(3 Pt 1):565-573.
- Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. (2007). Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. 15(9):943-949.
- Chai JH, Locke DP, Greally JM, Knoll JH, Ohta T, Dunai J, et al. (2003). Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 73(4):898-925.
- Doornbos M, Sikkema-Raddatz B, Ruijvenkamp CA, Dijkhuizen T, Bijlsma EK, Gijsbers AC, et al. (2009). Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur J Med Genet. 52(2-3):108-115.
- van der Zwaag B, Staal WG, Hochstenbach R, Poot M, Spierenburg HA, de Jonge MV, et al. (2010). A co-segregating microduplication of chromosome 15q11.2 pinpoints two risk genes for autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 153B(4):960-966.
- Burnside RD, Pasion R, Mikhail FM, Carroll AJ, Robin NH, Youngs EL, et al. (2011). Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 130(4):517-528.
- von der Lippe C, Rustad C, Heimdal K, Rødningen OK. (2011). 15q11.2 microdeletion - seven new patients with delayed development and/or behavioural problems. Eur J Med Genet. 54(3):357-660.
- Abdelmoity AT, LePichon JB, Nyp SS, Soden SE, Daniel CA, Yu S. (2012). 15q11.2 proximal imbalances associated with a diverse array of neuropsychiatric disorders and mild dysmorphic features. J Dev Behav Pediatr. 33(7):570-576.
- De Wolf V, Brison N, Devriendt K, Peeters H. (2013). Genetic counseling for susceptibility loci and neurodevelopmental disorders: the del15q11.2 as an example. Am J Med Genet A. 161A(11):2846-2854.
- Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, Boute O, et al. (2015). 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet. 58(3):140-147.
- Picinelli C, Lintas C, Piras IS, Gabriele S, Sacco R, Brogna C, et al. (2016). Recurrent 15q11.2 BP1-BP2 microdeletions and microduplications in the etiology of neurodevelopmental disorders. Am J Med Genet B Neuropsychiatr Genet. 171(8):1088-1098.
- Mohan KN, Cao Y, Pham J, Cheung SW, Hoffner L, Ou ZZ, et al. (2019). Phenotypic association of 15q11.2 CNVs of the region of breakpoints 1-2 (BP1-BP2) in a large cohort of samples referred for genetic diagnosis. J Hum Genet. 64(3):253-255.
- Benítez-Burraco A, Barcos-Martínez M, Espejo-Portero I, Jiménez-Romero S. (2017). Variable Penetrance of the 15q11.2 BP1-BP2 Microduplication in a Family with Cognitive and Language Impairment. Mol Syndromol. 8(3):139-147.
- Goytain A, Hines RM, El-Husseini A, Quamme GA. (2007). NIPA1(SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter. J Biol Chem. 282(11):8060-8068.
- Goytain A, Hines RM, Quamme GA. (2008). Functional characterization of NIPA2, a selective Mg2+ transporter. Am J Physiol Cell Physiol. 295(4):C944-C953.
- Reed JA, Wilkinson PA, Patel H, Simpson MA, Chatonnet A, Robay D, et al. (2005). A novel NIPA1 mutation associated with a pure form of autosomal dominant hereditary spastic paraplegia. Neurogenetics. 6(2):79-84.
- Jiang Y, Zhang Y, Zhang P, Sang T, Zhang F, Ji T, et al. (2012). NIPA2 located in 15q11.2 is mutated in patients with childhood absence epilepsy. Hum Genet. 131(7):1217-1224.
- Nowicki ST, Tassone F, Ono MY, Ferranti J, Croquette MF, Goodlin-Jones B, et al. (2007). The Prader-Willi phenotype of fragile X syndrome. J Dev Behav Pediatr. 28(2):133-138.
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, et al. (2012). De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 17(2):142-153.
- Chang X, Qu H, Liu Y, Glessner J, Hou C, Wang F, Li J, Slet al. (2019). Microduplications at the 15q11.2 BP1-BP2 locus are enriched in patients with anorexia nervosa. J Psychiatr Res. 113:34-38.
- Silva AI, Ulfarsson MO, Stefansson H, Gustafsson O, Walters GB, Linden DEJ, et al. (2019). Reciprocal White Matter Changes Associated With Copy Number Variation at 15q11.2 BP1-BP2: A Diffusion Tensor Imaging Study. Biol Psychiatry. 85(7):563–572.